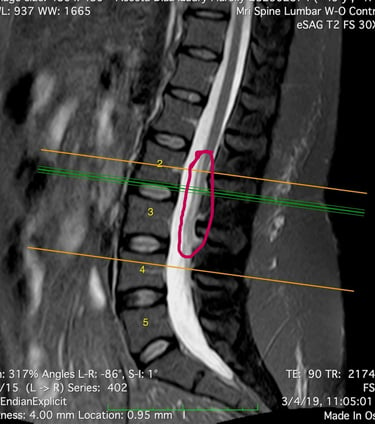
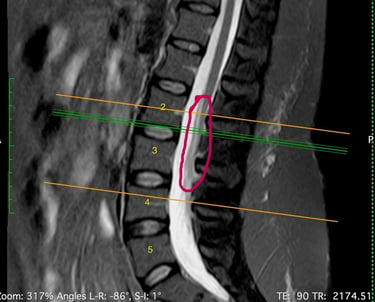
MEDULA ANCLADA
SIringomIelia
Klippel-Feil
La historia de nuestra fundadora
Todo empezó en el año 2019, cuando Audry resbaló por una escalera helada, resultando en una fractura del sacro.
Sus primeros síntomas fueron: dolor lumbar, dolores de cabeza, cuello y hombros. A finales del 2019 su doctor ordenó una resonancia magnética cervical luego de notar una anomalía en un ultrasonido del hombro derecho.
El resultado de la resonancia magnética fue el diagnóstico del "syrinx" o quiste en la médula espinal (Siringomielia) que se llena de líquido cefalorraquídeo, obstruyendo el canal al crecer. El radiólogo también notó la fusión de dos vértebras cervicales (Klippel-Feil), lo cual la dirigió a ver un neurocirujano, quien dijo no tener la experiencia necesaria para tratar esta enfermedad pero aseguró que los síntomas no eran causados por este diagnóstico.
La lista de síntomas continuaba creciendo (debilidad, adormecimiento, mareos, palpitaciones y latidos irregulares) y también el numero de doctores que visitaba tratando de conseguir respuestas; fue así como decidió consultar con un neurocirujano de los Estados Unidos, en julio del 2021, quien confirmó los tres diagnósticos: Siringomielia, Síndrome del Cordón Anclado y Síndrome de Klippel-Feil. Este último fue identificado en unas imágenes de resonancia magnética del ano 2019, las cuales habían sido vistas por varios doctores incluyendo radiólogo, ortopeda, neurocirujano y médico de familia, pero ninguno había podido identificar el defecto.
Al inicio del 2023, ella había visto alrededor de 20 doctores en Canada, había estado en la emergencia de los hospitales al menos una docena de veces y se había mudado a otra provincia; y fue en Alberta donde finalmente encontró un Genetista en Canadá que confirmó las tres enfermedades y, además, ordenó una prueba de micromatriz cromosómica, cuyo resultado arrojó una deleción en el cromosoma 15, llamada 15q 11.2, que es de rara incidencia y puede ser responsable de varias condiciones neurológicas.
Audry cuenta la historia completa en su primer libro que ya está a la venta en Amazon y posteriormente publicado en Inglés y Francés.
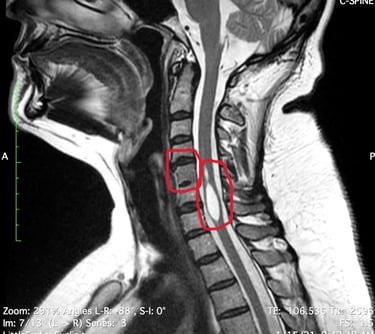
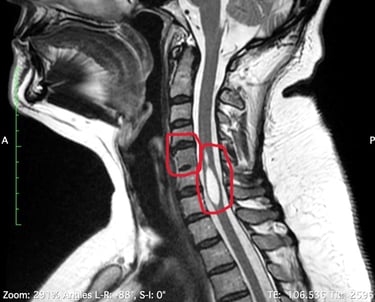
Demos un vistazo a estas enfermedades para tener mejor entendimiento de la causa, sintomas y tratamiento.
Informacion obtenida en la pagina oficial del National Institute of Health de los Estados Unidos (www.nih.gov).
SIRINGOMIELIA
La siringomielia es un trastorno en el cual se forma un quiste llamado siringe lleno de líquido cefalorraquideo dentro de la médula espinal. Con el tiempo, esta siringe se expande y se alarga lesionando la médula espinal. La incidencia de esta enfermedad es de 8.4 casos por 100,000 habitantes de los Estados Unidos.
Como la médula espinal conecta el cerebro con los nervios de las extremidades, este daño puede causar dolor, debilidad y rigidez en la espalda, los hombros, los brazos o las piernas. Los síntomas varían de una persona a otra. Otros síntomas pueden incluir dolores de cabeza y una pérdida de la sensibilidad al frío y al calor extremos, especialmente en las manos. Los signos del trastorno tienden a desarrollarse lentamente, aunque puede aparecer repentinamente al toser o hacer un esfuerzo. Si no se trata quirúrgicamente, la siringomielia puede producir debilidad progresiva en los brazos y las piernas, pérdida de la sensibilidad de la mano y dolor crónico intenso. En la mayoría de los casos, el trastorno está relacionado con una anomalía congénita del cerebro llamada malformación de Chiari, que hace que el tejido cerebral sobresalga de su ubicación normal en la parte posterior de la cabeza y en la porción cervical o del cuello del canal espinal. La siringomielia también puede ocurrir como una complicación de trauma, inflamación, lesión de la médula espinal, hemorragia, tumores de la médula espinal u otras afecciones. Los síntomas pueden aparecer meses o incluso años después de la lesión inicial, comenzando con dolor, debilidad y deterioro sensorial que se origina en el lugar del trauma. Algunos casos de siringomielia son hereditarios, aunque esto es poco común.
Los síntomas de la siringomielia congénita generalmente comienzan en la adultez temprana. Si no se trata quirúrgicamente (cuando es necesario), la siringomielia a menudo lleva a una debilidad progresiva de los brazos y las piernas, pérdida de la sensibilidad de la mano y dolor crónico intenso. Los síntomas pueden empeorar al hacer un esfuerzo o cualquier actividad que cause que fluctúe la presión del líquido cefalorraquídeo. Algunas personas pueden tener largos períodos de estabilidad. La cirugía da como resultado la estabilización o una mejoría modesta en los síntomas para la mayoría de las personas. El retraso en el tratamiento puede provocar una lesión irreversible en la médula espinal.
Si no hay síntomas, la siringomielia generalmente no se trata y un neurólogo o neurocirujano hace seguimiento al paciente. La cirugía generalmente se recomienda para personas con siringomielia sintomática o progresiva.
SINDROME DE KLIPPEL-FEIL
Es un trastorno poco común en el que dos o más vértebras del cuello se fusionan desde el nacimiento. Las personas que viven con el síndrome de Klippel-Feil pueden tener un cuello corto, movimiento limitado y dolor. Se estima que este síndrome ocurre en aproximadamente 1 de cada 40.000 a 42.000 recién nacidos en todo el mundo.
Los síntomas más comunes del síndrome de Klippel-Feil incluyen:
Cuello corto y posibilidad de tener una línea de cabello baja en la parte posterior de la cabeza. Flexibilidad y movimiento limitados, que afectan la cara, el cuello, la parte superior del cuerpo y la espalda. Dolor: las vértebras fusionadas pueden causar daño a los nervios y dolor en la cabeza, el cuello o la espalda. Pérdida de audición: las señales sonoras tienen dificultades para llegar a la parte derecha del oído o del cerebro.
Las personas con una determinada mutación genética tienen un mayor riesgo de desarrollar el trastorno. Las mutaciones en los genes GDF6 (factor de diferenciación de crecimiento 6) o GDF3 (factor de diferenciación de crecimiento 3) pueden causar el trastorno. Los médicos suelen diagnosticar el síndrome de Klippel-Feil en el momento del nacimiento o cerca de él mediante observación. Se pueden realizar pruebas para saber si el trastorno es leve o grave. Actualmente, los médicos tratan el síndrome de Klippel-Feil con cirugía para corregir los huesos del cuello. Las personas con síndrome de Klippel-Feil deben evitar actividades que puedan dañar el cuello. Es posible que las personas con síndrome de Klippel-Feil lleven una vida normal con el tratamiento y la atención adecuados.
El pronóstico depende de la gravedad del caso y de la cantidad de afecciones relacionadas que tenga la persona. Desafortunadamente, el síndrome de Klippel-Feil es una enfermedad rara y la mayoría de los casos ocurren sin una causa conocida. Esto significa que a menudo no se conoce mucha información sobre el trastorno. Los médicos e investigadores no ven suficientes pacientes con síndrome de Klippel-Feil.
SINDROME DE MEDULA ANCLADA
El síndrome de la médula espinal anclada (TSCS) es un trastorno del sistema nervioso causado por un tejido que se adhiere a la médula espinal y limita el movimiento de la médula espinal. Aún se desconoce la incidencia del trastorno en la población general.
Las inserciones de tejido pueden estar presentes desde el nacimiento en la base de la médula espinal (conocida como cono medular) o pueden desarrollarse cerca del sitio de una lesión en la médula espinal. Pueden hacer que la médula espinal se estire de forma anormal. El curso del trastorno es progresivo, lo que significa que empeora con el paso del tiempo. En los niños, los síntomas pueden incluir: Lesiones, zonas peludas, hoyuelos o tumores grasos en la parte baja de la espalda. Deformidades del pie y la columna. Debilidad en las piernas Dolor en la espalda baja Escoliosis Pérdida del control de los intestinos y la vejiga. El TSCS en niños parece ser causado por un crecimiento incorrecto del tubo neural mientras el feto se desarrolla y está relacionado con el trastorno llamado espina bífida. El TSCS puede no ser diagnosticado hasta la edad adulta. Los síntomas entonces pueden incluir:
Dolor
Problemas sensoriales y motores.
Pérdida del control de los intestinos y la vejiga
Estos síntomas tardíos están relacionados con la tensión que se ejerce sobre la médula espinal a lo largo del tiempo. La tensión puede empeorar durante los deportes o el embarazo, o puede deberse a que la columna vertebral se estrecha (un proceso conocido como estenosis) a medida que la persona envejece. El anclaje también puede desarrollarse después de una lesión de la médula espinal. El tejido cicatricial de la lesión puede bloquear el flujo de líquidos alrededor de la médula espinal. La presión de los líquidos puede provocar la formación de quistes en la médula espinal, una afección llamada siringomielia. Esto puede provocar una pérdida adicional de movimiento, entumecimiento, dolor u otros síntomas como aumento del ritmo cardíaco, presión arterial o problemas para respirar. Las imágenes por resonancia magnética se utilizan a menudo para evaluar a personas con síntomas de TSCS. Los médicos pueden utilizar imágenes por resonancia magnética para diagnosticar dónde se encuentra el anclaje, si la base de la médula espinal de un paciente (el cono medular) está más baja de lo normal o si un tumor o una masa grasa (conocida como lipoma) está causando los síntomas de TSC. En niños con TSCS, generalmente se recomienda la cirugía temprana para prevenir un mayor deterioro neurológico. Si no se recomienda la cirugía, se pueden cortar las raíces nerviosas de la médula espinal para aliviar el dolor. En adultos, la cirugía para liberar (desanudar) la médula espinal puede reducir el tamaño y el mayor desarrollo de los quistes en la médula. La cirugía también puede restaurar alguna función o aliviar otros síntomas. Tanto para niños como para adultos, otras formas de tratamiento pueden ayudar a aliviar los síntomas del TSCS.
DELECION 15q 11.2
(Información encontrada en el sitio web oficial de "Unique" www.rarechromo.org)
Una deleción 15q11.2 es una variación genética muy rara en la que falta una pequeña parte del cromosoma 15. La eliminación se encuentra en un lugar llamado q11.2. Debido a que la pieza que falta es muy pequeña, a veces la verás como microdeleción.
Lo que sabemos sobre las microdeleciones 15q11.2 proviene del estudio de personas que tienen un motivo para realizarse una prueba genética. La razón podría ser un retraso en el desarrollo, un comportamiento inusual o un problema de salud, o tal vez se haya encontrado la microdeleción 15q11.2 en alguien más de su familia. Esto nos da una muestra sesgada. Si buscáramos la microdeleción 15q11.2 en la población general tendríamos una muestra imparcial, pero es muy difícil de hacer. Esto significa que por el momento no podemos estar seguros del efecto de una microdeleción 15q11.2.
Todavía queda mucho por aprender, pero esta guía contiene la mejor información que tenemos hasta la fecha. Las características de las personas con una microdeleción 15q11.2 varían ampliamente, incluso entre miembros de una misma familia. Las personas pueden tener retrasos en el desarrollo, dificultades de aprendizaje y problemas de conducta. Sin embargo, muchas personas con microdeleción no tienen dificultades físicas, de aprendizaje o de comportamiento aparentes. Debido a que sólo se ha identificado un número muy pequeño de personas, todavía no podemos estar seguros de cuál es la gama completa de posibles efectos de la microdeleción. Además, las características varían, incluso entre miembros de una misma familia. No afectan a todo el mundo y en cualquier individuo pueden ser más o menos evidentes. Las características más comunes son: Los niños pueden necesitar apoyo con el aprendizaje. La cantidad de apoyo que necesita cada niño variará, aunque la mayoría se beneficia de servicios de apoyo para necesidades especiales. Trastornos emocionales y de conducta, incluido el trastorno por déficit de atención con hiperactividad y/o autismo en algunos niños. Retraso del habla en algunos niños. A veces la microdeleción 15q11.2 puede ser silenciosa. Algunos padres de niños con una microdeleción 15q11.2 tienen la misma microdeleción pero no tienen ninguna característica inusual obvia ni retraso en el desarrollo. El efecto de algunos trastornos genéticos sobre el desarrollo, la salud y el comportamiento varía desde apenas perceptible hasta obvio y grave.
Las enfermedades que padece
info@myrareuniverse.org
© 2023 All rights reserved My Rare Universe Foundation
P.O.Box 82113
Edmonton RPO Yellowbird, AB T6J 7E6
Edmonton, Alberta CANADA