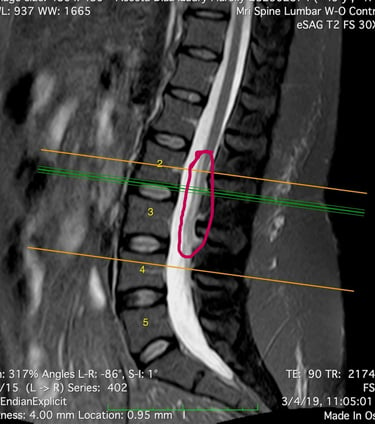
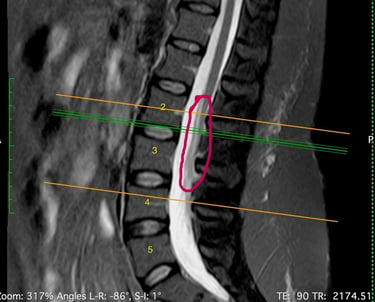
MEDULA ANCLADA
SIringomIelia
Klippel-Feil
L'histoire de notre fondateur.
Tout a commencé en 2019 lorsqu'Audry a glissé dans un escalier verglacé, se fracturant le sacrum.
Ses premiers symptômes comprenaient des douleurs lombaires, des maux de tête et des douleurs à la nuque et aux épaules. Fin 2019, son médecin a prescrit une IRM cervicale après avoir constaté une anomalie lors d'une échographie de son épaule droite.
L'IRM a révélé une syringomyélie, un kyste de la moelle épinière qui se remplit de liquide céphalo-rachidien et obstrue le canal rachidien en grossissant. Le radiologue a également constaté la fusion de deux vertèbres cervicales (syndrome de Klippel-Feil), ce qui l'a conduite à consulter un neurochirurgien. Ce dernier a déclaré ne pas être en mesure de traiter cette affection, mais l'a assurée que ses symptômes n'étaient pas liés à ce diagnostic.
La liste des symptômes n'a cessé de s'allonger (faiblesse, engourdissements, vertiges, palpitations et arythmie cardiaque), tout comme le nombre de médecins qu'elle a consultés pour tenter de comprendre. C'est alors qu'en juillet 2021, elle a décidé de consulter un neurochirurgien aux États-Unis, qui a confirmé les trois diagnostics : syringomyélie, syndrome de la moelle attachée et syndrome de Klippel-Feil. Ce dernier avait été identifié sur des IRM réalisées en 2019, qui avaient été examinées par plusieurs médecins, dont un radiologue, un orthopédiste, un neurochirurgien et un médecin généraliste, mais aucun n'avait pu déceler l'anomalie.
Au début de 2023, elle avait consulté une vingtaine de médecins au Canada, s'était rendue aux urgences hospitalières au moins une douzaine de fois et avait déménagé dans une autre province ; c'est en Alberta qu'elle a finalement trouvé un généticien au Canada qui a confirmé les trois maladies et, de plus, a prescrit un test de microarray chromosomique, dont le résultat a montré une délétion sur le chromosome 15, appelée 15q11.2, qui est rare et peut être responsable de plusieurs affections neurologiques.
Audry raconte toute l'histoire dans son premier livre, déjà disponible sur Amazon et qui sera publié ultérieurement en anglais et en français.
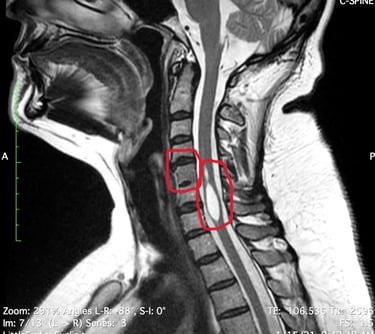
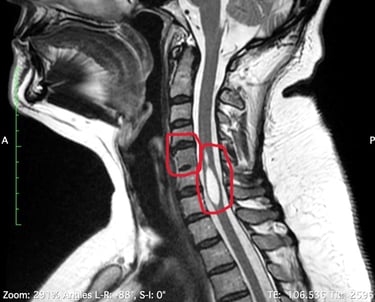
Examinons ces maladies afin de mieux comprendre leurs causes, leurs symptômes et leurs traitements.
Information tirée du site web officiel des Instituts nationaux de la santé des États-Unis (www.nih.gov).
SYRINGOMYÉLIE
La syringomyélie est un trouble dans lequel un kyste rempli de liquide céphalorachidien, appelé syrinx, se forme à l'intérieur de la moelle épinière. Avec le temps, ce syrinx s'élargit et s'allonge, endommageant la moelle épinière. L'incidence de cette maladie est de 8,4 cas pour 100 000 habitants aux États-Unis.
Comme la moelle épinière relie le cerveau aux nerfs des membres, ces dommages peuvent provoquer des douleurs, de la faiblesse et de la raideur dans le dos, les épaules, les bras ou les jambes. Les symptômes varient d'une personne à l'autre. D'autres symptômes peuvent inclure des maux de tête et une perte de sensibilité au froid et à la chaleur extrêmes, en particulier dans les mains. Les signes du trouble ont tendance à se développer lentement, bien qu'ils puissent apparaître soudainement lors d'une toux ou d'un effort. Si elle n'est pas traitée chirurgicalement, la syringomyélie peut entraîner une faiblesse progressive des bras et des jambes, une perte de sensibilité des mains et des douleurs chroniques intenses.
Dans la majorité des cas, le trouble est lié à une anomalie congénitale du cerveau appelée malformation de Chiari, qui provoque une saillie du tissu cérébral hors de son emplacement normal à l'arrière de la tête et dans la partie cervicale du canal rachidien. La syringomyélie peut également survenir comme complication d'un traumatisme, d'une inflammation, d'une lésion de la moelle épinière, d'une hémorragie, de tumeurs de la moelle épinière ou d'autres affections. Les symptômes peuvent apparaître des mois, voire des années après la lésion initiale, commençant par de la douleur, de la faiblesse et une détérioration sensorielle à l'endroit du traumatisme. Certains cas de syringomyélie sont héréditaires, bien que cela soit rare.
Les symptômes de la syringomyélie congénitale commencent généralement au début de l'âge adulte. Si elle n'est pas traitée chirurgicalement (lorsque cela est nécessaire), la syringomyélie mène souvent à une faiblesse progressive des bras et des jambes, une perte de sensibilité des mains et des douleurs chroniques intenses. Les symptômes peuvent s'aggraver lors d'un effort ou de toute activité provoquant une fluctuation de la pression du liquide céphalorachidien. Certaines personnes peuvent connaître de longues périodes de stabilité. La chirurgie permet une stabilisation ou une amélioration modeste des symptômes pour la plupart des patients. Un retard de traitement peut entraîner des lésions irréversibles de la moelle épinière.
En l'absence de symptômes, la syringomyélie n'est généralement pas traitée et un neurologue ou un neurochirurgien assure le suivi du patient. La chirurgie est généralement recommandée pour les personnes présentant une syringomyélie symptomatique ou progressive.
SYNDROME DE KLIPPEL-FEIL
Il s'agit d'un trouble rare dans lequel deux vertèbres du cou, ou plus, sont fusionnées dès la naissance. Les personnes vivant avec le syndrome de Klippel-Feil peuvent avoir un cou court, une mobilité limitée et des douleurs. On estime que ce syndrome survient chez environ 1 nouveau-né sur 40 000 à 42 000 dans le monde.
Les symptômes les plus fréquents du syndrome de Klippel-Feil incluent :
Cou court et possibilité d'avoir une implantation des cheveux basse à l'arrière de la tête.
Flexibilité et mouvements limités, affectant le visage, le cou, le haut du corps et le dos.
Douleur : les vertèbres fusionnées peuvent causer des dommages nerveux et des douleurs à la tête, au cou ou au dos.
Perte d'audition : les signaux sonores ont des difficultés à atteindre la partie droite de l'oreille ou du cerveau.
Les personnes porteuses d'une certaine mutation génétique présentent un risque plus élevé de développer ce trouble. Des mutations dans les gènes GDF6 (facteur de différenciation de croissance 6) ou GDF3 (facteur de différenciation de croissance 3) peuvent être à l'origine de la maladie. Les médecins diagnostiquent généralement le syndrome de Klippel-Feil au moment ou peu après la naissance par simple observation. Des examens peuvent être réalisés pour déterminer si le trouble est léger ou grave. Actuellement, les médecins traitent le syndrome de Klippel-Feil par la chirurgie pour corriger les os du cou. Les personnes atteintes doivent éviter les activités susceptibles de blesser le cou. Il est possible de mener une vie normale avec un traitement et des soins appropriés.
Le pronostic dépend de la gravité du cas et du nombre d'affections associées. Malheureusement, le syndrome de Klippel-Feil est une maladie rare et la plupart des cas surviennent sans cause connue. Cela signifie que les informations disponibles sur ce trouble sont souvent limitées. Les médecins et les chercheurs ne voient pas suffisamment de patients atteints du syndrome de Klippel-Feil.
SYNDROME DE LA MOELLE ATTACHÉE
Le syndrome de la moelle attachée (ou moelle épinière ancrée - Tethered Spinal Cord Syndrome ou TSCS) est un trouble du système nerveux causé par des tissus qui adhèrent à la moelle épinière, limitant ainsi sa mobilité. L'incidence exacte de ce trouble dans la population générale reste encore inconnue.
Ces attaches tissulaires peuvent être présentes dès la naissance à la base de la moelle épinière (appelée cône médullaire) ou se développer à proximité d'un site de lésion médullaire. Elles provoquent un étirement anormal de la moelle. L'évolution du trouble est progressive, ce qui signifie qu'il s'aggrave avec le temps.
Chez les enfants, les symptômes peuvent inclure :
Lésions, zones pileuses (poils), fossettes ou tumeurs graisseuses dans le bas du dos.
Déformations des pieds et de la colonne vertébrale.
Faiblesse dans les jambes.
Douleurs lombaires.
Scoliose.
Perte de contrôle des intestins et de la vessie (incontinence).
Chez l'enfant, le syndrome semble être causé par un développement anormal du tube neural lors de la croissance fœtale et est étroitement lié au spina-bifida.
Le diagnostic peut ne pas être posé avant l'âge adulte. Les symptômes incluent alors :
Douleur.
Troubles sensoriels et moteurs.
Perte de contrôle des intestins et de la vessie.
Ces symptômes tardifs sont liés à la tension exercée sur la moelle épinière au fil du temps. Cette tension peut s'aggraver lors de la pratique de sports, pendant la grossesse, ou en raison du rétrécissement du canal rachidien (sténose) avec l'âge.
L'attache peut également se développer après une lésion de la moelle épinière. Le tissu cicatriciel peut alors bloquer la circulation des fluides autour de la moelle. La pression des fluides peut provoquer la formation de kystes, une affection appelée syringomyélie. Cela peut entraîner une perte de mobilité supplémentaire, des engourdissements, des douleurs ou d'autres symptômes tels qu'une accélération du rythme cardiaque, une tension artérielle instable ou des difficultés respiratoires.
Diagnostic et traitement :
L'IRM est fréquemment utilisée pour évaluer les personnes présentant des symptômes. Les médecins l'utilisent pour localiser l'attache, vérifier si le cône médullaire est plus bas que la normale, ou si une masse graisseuse (lipome) provoque les symptômes.
Chez les enfants : Une chirurgie précoce est généralement recommandée pour prévenir toute détérioration neurologique supplémentaire. Si la chirurgie n'est pas préconisée, les racines nerveuses de la moelle peuvent parfois être sectionnées pour soulager la douleur.
Chez les adultes : La chirurgie de libération médullaire peut réduire la taille et stopper le développement des kystes, restaurer certaines fonctions ou soulager d'autres symptômes.
Pour les enfants comme pour les adultes, d'autres formes de traitement peuvent aider à soulager les symptômes associés.
DÉLÉTION 15q11.2
(Informations issues du site officiel de Unique www.rarechromo.org)
Une délétion 15q11.2 est une variation génétique très rare dans laquelle une petite partie du chromosome 15 est manquante. La perte se situe à un emplacement appelé q11.2. Comme le segment manquant est très petit, on parle parfois de microdélétion.
Ce que nous savons des microdélétions 15q11.2 provient de l'étude de personnes ayant une raison de passer un test génétique. Cette raison peut être un retard de développement, un comportement inhabituel, un problème de santé, ou parce que la microdélétion 15q11.2 a été identifiée chez un autre membre de la famille. Cela nous donne un échantillon biaisé. Si nous recherchions la microdélétion 15q11.2 dans la population générale, nous aurions un échantillon impartial, mais c'est très difficile à réaliser. Cela signifie qu'actuellement, nous ne pouvons pas être certains de l'effet systématique d'une microdélétion 15q11.2.
Il reste encore beaucoup à apprendre, mais ce guide contient les meilleures informations dont nous disposons à ce jour. Les caractéristiques des personnes porteuses d'une microdélétion 15q11.2 varient considérablement, même au sein d'une même famille. Certaines personnes peuvent présenter des retards de développement, des difficultés d'apprentissage et des troubles du comportement. Cependant, de nombreuses personnes porteuses de cette microdélétion ne présentent aucune difficulté physique, d'apprentissage ou de comportement apparente.
Comme seul un petit nombre de personnes a été identifié, nous ne connaissons pas encore toute la gamme des effets possibles. De plus, les caractéristiques varient et ne touchent pas tout le monde ; chez chaque individu, elles peuvent être plus ou moins évidentes. Les caractéristiques les plus communes sont :
Soutien à l'apprentissage : Les enfants peuvent avoir besoin d'aide. Le niveau de soutien varie, bien que la plupart bénéficient de services adaptés aux besoins spécifiques.
Troubles émotionnels et du comportement : Incluant le trouble du déficit de l'attention avec ou sans hyperactivité (TDAH) et/ou l'autisme chez certains enfants.
Retard de langage : Observé chez certains enfants.
Parfois, la microdélétion 15q11.2 peut être « silencieuse ». Certains parents d'enfants porteurs ont la même microdélétion mais ne présentent aucune caractéristique inhabituelle évidente ni retard de développement. L'effet de certains troubles génétiques sur le développement, la santé et le comportement varie d'à peine perceptible à évident et grave.
Les maladies dont vous souffrez
info@myrareuniverse.org
© 2023 All rights reserved My Rare Universe Foundation
P.O.Box 82113
Edmonton RPO Yellowbird, AB T6J 7E6
Edmonton, Alberta CANADA